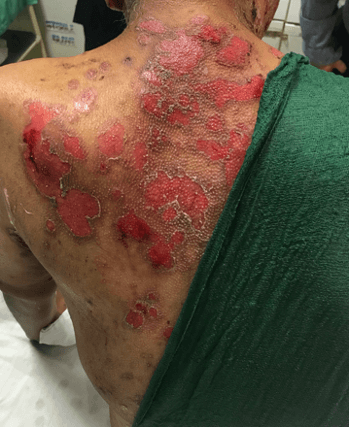
A 50-year-old woman presents with a painful, blistering rash affecting her face, limbs and trunk. She initially noticed painful blisters in her mouth, but these subsequently spread to the skin a few weeks later. The blisters, where present are large and flaccid. Most of the lesions, however, have sheared off by rubbing against her clothes and few intact lesions can be found. The rash is painful, but not itchy. A photo is shown below:
Show Answer
This patient has a diagnosis of pemphigus vulgaris. Pemphigus (which is derived from the Greek word for bubble ‘pemphix’) is a group of autoimmune disorders in which there is blistering of the skin and/or mucosal surfaces. It has an autoimmune basis with autoantibodies directed against cell surface antigens on keratinocytes (desmogleins 1 and 3). This results in loss of cell-to-cell adhesion and separation.
There are two major variants of pemphigus; pemphigus vulgaris and pemphigus foliaceous. Pemphigus herpetiformis, IgA pemphigus, paraneoplastic pemphigus and IgG/IgA pemphigus are rarer forms.
Pemphigus vulgaris (PV) is the most common subset, comprising around 70% of cases worldwide. The typical clinical features of PV include the following:
- Usually starts with painful blisters on the oral mucosa, which can occur weeks or months before skin lesions become evident
- Skin lesions are large and flaccid
- The affected skin is painful but rarely itchy
- Nikolsky sign is often present – the dislodgement of intact superficial epidermis by a shearing force
- Intact blisters may not be found
- PV is most common in patients between the ages of 40 and 60.
Pemphigus foliaceous (PF) is a relatively benign form of pemphigus. The typical clinical features of PF include the following:
- This presents with skin lesions only and the oral mucosa are spared
- Blisters usually appear initially on the face and scalp and then spread to the chest and back
- Blisters are typically itchy and painless
Question image used on licence from Shutterstock
Show Answer
Skin biopsy for histology and direct immunofluorescence is usually the investigation of choice. This shows intra-epidermal deposition of IgG between cells throughout the epidermis. ELISA to detect anti-desmoglein 1 and anti-desmoglein 3 autoantibodies can also be undertaken.
Show Answer
Treatment is aimed at reducing pain and symptoms and preventing complications like infection. High-dose corticosteroids are the first-line treatment, e.g. prednisolone 60 mg od. Steroids should not be tapered until most lesions (about 80%) have healed, to reduce the chances of a subsequent flare in disease activity. Steroid-sparing agents are sometimes used in an attempt to further reduce the dose, e.g. azathioprine, cyclophosphamide and dapsone. Rituximab given with intravenous immunoglobulin has also been successfully used in cases which resist standard treatments. Plasmapheresis can be used in very severe cases.
Show Answer
Secondary infection is a relatively common complication, particularly of skin lesions, and this may delay healing and increase scarring.
Untreated, the mortality associated with PV is around 75%. The use of corticosteroids and adjuvant drugs has reduced the mortality rate significantly to between 10-15%.
Header image used on licence from Shutterstock




very useful
Excellent
Very nice