
A phaeochromocytoma is a rare functional tumour that arises from chromaffin cells in the adrenal medulla. Extra-adrenal paragangliomas (extra-adrenal pheochromocytomas) are closely related, though less common, tumours that originate in the ganglia of the sympathetic nervous system. These tumours secrete catecholamines and cause a collection of symptoms and signs related to sympathetic nervous system hyperactivity.
The incidence is approximately 0.8 per 100,000 person-years and is estimated to be between 0.1-0.6% in patients with hypertension. The peak incidence occurs in the third and fourth decades of life.
Aetiology
The aetiology of phaeochromocytomas is not fully understood, but a significant genetic component is recognised, and genes have been identified as genomic biomarkers to aid in the identification of inherited disease. The majority are, however, thought to occur sporadically.
Phaeochromocytomas are known to occur in the following familial syndromes:
- Multiple endocrine neoplasia (MEN) syndromes
- Neurofibromatosis
- Von Hippel-Lindau (VHL) disease
- Sturge-Weber syndrome
- Familial phaeochromocytoma-paraganglioma syndrome
Ten per cent rule
The ‘ten per cent rule’ is a useful aide de memoir for phaeochromocytomas:
- 10% are bilateral (but 70% in familial cases)
- 10% are multiple
- 10% are not associated with hypertension
- 10% are extra-adrenal, (most commonly, in the organ of Zuckerkandl, located in the aortic bifurcation)
- 10% are malignant, (but the risk of malignancy in women is three times that for men)
- 10% occur in children (but 25-30% of children have extra-adrenal and/or bilateral tumours)
Clinical features
The most common presenting feature is hypertension, which can be sustained or paroxysmal.
Symptoms tend to be intermittent and can occur anything from several times a day to very infrequently. Over the clinical course of the disease, the symptoms tend to become more severe and increasingly frequent.
In addition to hypertension, the following clinical features may be present:
- Headache
- Profuse sweating
- Palpitations/tachycardia
- Tremor
- Fever
- Nausea and vomiting
- Anxiety and panic attacks
- Sense of ‘impending doom’
- Epigastric or flank pain
- Constipation
- Hypertensive retinopathy
- Postural hypotension (secondary to volume contraction)
- Cardiomyopathy
- Café au lait spots
Investigations
In patients with suspected phaeochromocytoma, the diagnosis can be confirmed by measuring elevated levels of metanephrines (catecholamine metabolites) in the blood or urine. Any of the following methods can be used:
- 24-hour urine collection for free catecholamines vanillylmandelic acid (VMA) and metanephrines
- High-performance liquid chromatography for catecholamines in plasma and/or urine
- Radioimmunoassay (RIA) for urinary/plasma metanephrines
After biochemical confirmation of phaeochromocytoma, the tumour should be located via one of the following imaging methods:
- CT scan – the first imaging modality to be used (overall sensitivity of 89%)
- MRI scan – the most sensitive modality for identification of the tumour and is very useful in extra-adrenal cases and metastatic disease (overall sensitivity of 98%)
- Nuclear medicine scan, e.g. MIBG scintigraphy – useful in cases that have been biochemically confirmed but CT or MRI does not show a tumour.
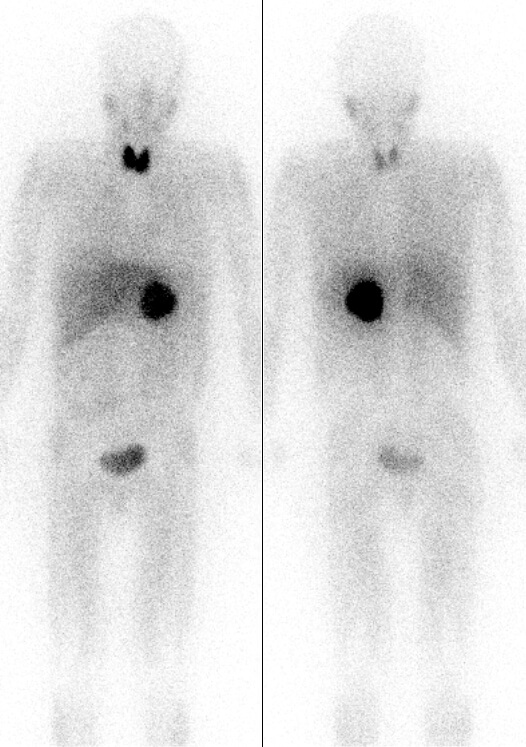
Pheochromocytoma localised by MIBG scintigraphy, image sourced from Wikipedia
Courtesy of the Drahreg01 CC BY-SA 3.0
Management
Surgical resection is the definitive treatment of choice, and if complete resection is achieved, without metastases, this usually results in the cure of hypertension.
Pre-operative medical management is essential as this significantly reduces the risk of intraoperative hypertensive crises. This is usually achieved through the use of a combination of non-competitive alpha-blockers (e.g. phenoxybenzamine) and beta-blockers. Alpha-blockade should be initiated first, at least 7-10 days before surgery to allow for expansion of blood volume. Once this has been achieved, beta-blockade can be commenced, which helps top control tachycardia and certain arrhythmias. If beta-blockade is initiated too early hypertensive crisis can be precipitated.
Genetic counselling should also take place, and associated conditions looked for and managed as appropriate.
Header image used on licence from Shutterstock
Thank you to the joint editorial team of www.plabprep.co.uk for this ‘Exam Tips’ post.




